This version of our Researcher Spotlight takes a slightly different, Q&A-style narrative, because Vijay’s tireless work has inspired a massive breakthrough in Sickle Cell Anemia treatment.
Let’s dig in!
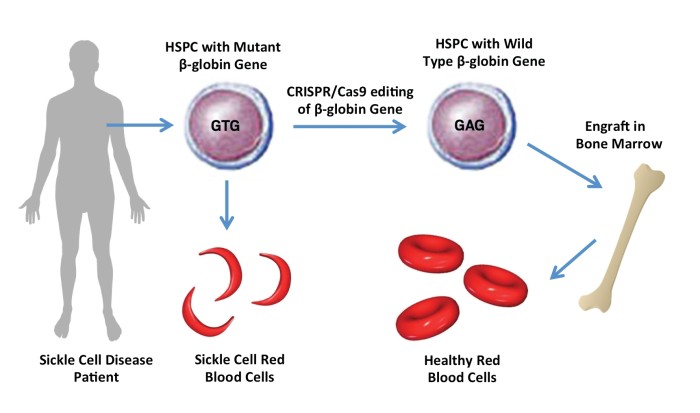
Q: We wanted to talk to you about the recently, December 2023, FDA approved gene therapy Casgevy, to treat transfusion-dependent beta thalassemia (TDT), a type of sickle cell disease. As we understand, this is the culmination of work you started during your PhD back in 2004, in Stuart Orkin’s lab. Could you please tell us a bit about what piqued your interest back then to work on this project?
A: Oh, well, maybe I’d answer that in a little bit of a different way, if that’s okay? – Because I think it’s a great question! – I think that I became very fascinated by the problem we were trying to address, which was: ‘how during human development is there this switch from a fetal form of hemoglobin that you express throughout much of gestation to the adult human’ – And we had known from decades of work, including work here at Boston Children’s Hospital, that if you can induce that fetal form of hemoglobin, or if you naturally just have more of the fetal hemoglobin, you actually can do much better if you have sickle cell disease or dialysis…and that was known for a long, long time. Yet how this process was regulated, was really unknown.
And I bring that up because, you know, when I entered my PhD I was like, “okay, I really want to understand this problem”, a little bit naively, but there was a lot known about it, and not very much known about the molecular basis of it.
When I started my PhD and wanted to address this problem, I went to my PhD advisor – He said, ‘“oh, you know, [he was sort of a little bit skeptical because he’d worked on this], decades ago…lots of people have kind of lost their careers doing this…but why don’t we go ahead”. And soon thereafter, he said, “why don’t you help me write a grant?” [And that grant renewal]…it actually got an almost perfect score…at the time it was like 110 or something. It was really well received. But I can tell you retrospectively [laughing] that every one of the aims of that grant proposal actually totally failed.
The [aims] failed in part because there were assumptions made about the system. And it turned out that the thing which really helped us make a lot of advances was advances in genomics and human genetics. And specifically, we were able to identify this association in the BCL11A gene through genome-wide association studies.
So I bring all that up because, as we think about the CDN and the value of emerging genomic approaches that are happening…to me, I see tremendous value because as I look back to what we ‘were able to do’, we had a question in mind – but couldn’t answer it – and it was really through advances in genomics and human genetics, that we could make any headway to understand it.
I say all of this because I think it’s a broader message that I hope will ring true many more times! That’s what excites me about all these advances in single cell biology…we now have an opportunity, I think in many ways, akin to what happened twenty-ish years ago, that will allow us to really start to make some of those important advances.
Q: Could you explain a little bit about what Sickle Cell disease is, and specifically transfusion-dependent beta thalassemia (TDT) is, how it works, and its prevalence?
A: At a basic level, both sickle cell disease and beta thalassemia, are actually the most common monogenic diseases in the world…because of selection for carriers..who have these mutations, because it prefers resistance to severe forms of malaria, it turns out. But really both of them occur in the same gene!..so, due to mutations. The adult beta hemoglobin molecule or HBbV is the gene, and it turns out that sickle cell disease, there’s a specific point mutation – that causes the hemoglobin, once it’s assembled as a protein to have a tendency in the deoxygenated state to polymerize and actually to form red cells. So you get these polymers of hemoglobin that form, and they deformed the red cells and cause [sort of] red cells to then stick in small blood vessels, blocking blood circulation, causing pain and organ damage as a result.
Thalassemia on contrast, is a condition also due to mutations in the adult beta hemoglobin molecule…but it’s really [moreso] due to reduced production of the adult beta hemoglobin molecule…so there, you have a production issue.
Really, the problems in beta thalassemia…to dive into that a little bit more, are not because of the deficiency of beta hemoglobin, but actually because of the imbalance between the other part of hemoglobin, the alpha hemoglobin and the beta hemoglobin – if you have too much of the free ‘alphas’, it turns out they actually precipitate themselves and cause this sort of precursors in the bone marrow…to die and not be produced effectively.
The reason I bring up that both of ‘these’ are showing these two problems…the adult beta hemoglobin molecule, it turns out that if you can get rid of, or turn down the amount of adult beta hemoglobin, you can help with both of these conditions! And the way you naturally do that is, during gestation you have this fetal form of hemoglobin, and that is a beta-like hemoglobin molecule, but it substitutes for beta hemoglobin – so nature has sort of devised this ‘way’, and it turns out that children with sickle cell disease or thalassemia don’t manifest into latent infancy with symptoms typically, because they’re protected while this fetal hemoglobin is ‘on’ – so, nature has sort of shown us how to do this! We just didn’t know what the molecular regulation of that process was.
Q: Why do we need these two Beta Hemoglobin Molecules/why don’t we just keep one for the entire time?
A: I think it’s a really great question! Actually, most mammals had done just that. Outside of old world primates, almost every other mammal has just a [one] adult hemoglobin – and no switching process! Probably some of it has to do with the fact that fetal hemoglobin has a higher oxygen affinity, so it facilitates a transplacental oxygen transport. But it turns out, for example, that if you measure mouse red cells – mice do a fine job of transferring transplacentally oxygen – there’s just other adaptations. you can have to enable that. I bring this up because it turns out, there are humans naturally with mutations or deletions that cause high levels of fetal hemoglobin, and even if you have 98% fetal hemoglobin, you can give birth just fine…So there’s ways that you can adapt and get around this, but of course, evolution doesn’t care about the individual that cares about thousands of individuals. And so maybe it would help people over time to have this kind of adaptation. But it’s a really great question because, ‘where and how it evolved’, is something that we don’t fully understand.
Q: What is the main role of BCL11A?
A: BCL11A really serves in some ways as a rheostat to regulate fetal hemoglobin. So what we know so far is, it acts as a transcription factor – and when we uncovered BCL11A’s role in fetal hemoglobin switching, it turned out, it was really well studied for its role in B lymphocyte development and its role in neural development.
So people had characterized it as an important transcription factor for both of these processes. And its role in red cell production was just not at all appreciated, right? It wasn’t one of the key regulators of red cell production canonically…I think of it as an accent, because as soon as we found out, we said ‘alright, let’s turn it down, see what happens at the time etc…’, with sort of the antiquated siRNA or shRNA approaches… immediately, we could see this result where, robustly, you reduced fetal hemoglobin. I mean, it was remarkable. And the reason I say it was remarkable is because we had been trying all these other factors that we had had hypotheses about and all of them had failed, right? – so we knew the system was working as we expected…and yet, we never hit upon something that had this effect.
Q: A little bit later in 2015, Daniel Bauer published a paper where he found out about the enhancer that promotes the expression of BCL11A, can you tell us a bit about your experience as it relates?
When did you think about using the CRISPR-CAS9 system as a mode of therapy?
A: Right, yes, that is a very interesting story!
They had been looking for where the genetic association was. We knew it was in the BCL11A gene, but it was in a non-coding sequence. It turned out that it was mapped to a region that contained this enhancer. The funny part of the story is they identified a large PANA10 KV region that included the variation. But, to this day, we still do not understand how these variants act and they only bring this up because I still think that there’s more biology to learn…a lot to learn about how this variation acts, and things that we’re trying to study related to this. Because they identified an enhancer, they actually started to go into and use CRISPR tiling approaches to actually map the most active parts of that in cancer. One of these reasons, which is what they published in the 2015 paper from Canberra and colleagues, was actually the really active region that was sort of the key for allowing expression – and if you edited that, One of these reasons, which is what they published in the 2015 paper from Canberra and colleagues. It was actually the really active region that was sort of the key for allowing expression – and if you edited that, which is essentially what KAF-JV does, then you can nicely turn down BCL11A levels.